571z Analyzing and Overcoming of Baculovirus Mutants Obtained from Passaging In Insect Cell Culture. A Potential Approach to Low-Cost and Large-Scale Baculovirus Production
Introduction: Biopesticides/biological controls are extremely useful in overcoming the significant disadvantages of chemical pesticides. However, the current production cost of a potential biopesticide, such as baculovirus (which kills only targeted insects and is benign to human health and environment), is much higher than that of chemical pesticides. Moreover, the baculovirus insect-cell expression system is widely used for the expression of recombinant proteins in eukaryotic environment. Specifically, the proteins expressed in this system often show biological activities similar to eukaryotic species due to the proper folding and post-translational modification. Hence, the establishment of large scale continuous production of baculovirus-expressed proteins can be of great importance in the area of pharmaceuticals, vaccines (subunit) and diagnostics. But a low cost, large-scale production of the baculovirus Autographa californica multiple nucleopolyhedrovirus (AcMNPV) using a continuous insect cell culture is greatly hindered by undesirable mutations in the baculovirus genome, such as few polyhedra (FP) and defective interfering particle (DIP) mutants. Accumulations of these mutants affect the normal functioning of the virus and thereby the quality and quantity of the product virus and recombinant protein is greatly compromised. Overcoming such mutations is an extremely important step in enabling a cheaper large-scale production of baculovirus in insect cell culture. In this context, our primary goal is to overcome the FP mutation by genetic modification of the baculovirus or the host cell. In addition, we will examine whether FP mutants are a necessary precursor of the deletion mutants, namely, defective interfering particle (DIP) mutants by characterization of the mutant formation during continuous passaging.
Background on budded virus and polyhedra: Baculoviruses are a family of invertebrate viruses having a double-stranded circular DNA genome. They have two morphological forms: the budded virus (BV) and the occluded virus (OV). The BV's are encased in a membrane and bud through the cellular membrane to multiply within an infected insect. On the other hand, OV's are enveloped singly or multiply in an occlusion body within the cell's nucleus; OV's are embedded in polyhedrin protein matrix and called “polyhedra” which can be used as a pesticide due to its host specificity.
Background on FP mutation and DIP mutation: The FP mutation generally results from the insertion of host insect cell DNA sequences, known as transposons, into the baculovirus fp25k gene at TTAA target sites (Fraser et al., 1983). FP mutation/FP phenotype in baculovirus is characterized by reduced virion occlusion, fewer polyhedra/cell, fewer or no viruses/polyhedra and increased budded virus production and altered intranuclear envelopment. On the other hand, the DIP mutants result from deletion of almost 40% genome from the WT virus and it causes significant reduction in viral infectivity as well as the level of recombinant protein production (Kool et al., 1991).
Genetically engineered “stabilized virus” and “passaging experiment”: In order to overcome the FP mutation resulting from the insertion of transposons in the fp25k gene, the wild type (WT) AcMNPV is modified by removing the TTAA sites from fp25k gene using template directed ligation and PCR. This new engineered virus is expected to be more stable during passaging in cell culture and named as “stabilized virus”. A “passaging experiment” was performed where three types of viruses, WT, stabilized and delta (fp25k gene deleted, used as negative control) AcMNPV, were passaged upto the 32 times in Spodoptera frugiperda (Sf-21) cells. As the quality of the product virus in cell culture is dependent on the “passaging event”, a detailed analysis of the mutant formation was performed with the passaged viruses to evaluate the effectiveness of the stabilized virus during passaging.
Immunofluorescent assay and western blot (Fig 1): An immunofluorescent assay was designed to detect the infected cells expressing FP25K in a qualitative and quantitative manner. It was found that the fraction of cells expressing FP25K protein was higher for the stabilized virus upto passage 16. For further verification, western blotting was performed with the infected cell lysates using FP25K antibody. It was shown that with passaging, the degradation of functional FP25K protein occurs at a lower rate in case of the stabilized virus.
Cell growth/infection pattern: The host cell growth curve and infection pattern was compared for the two types of viruses (passage 1, 16 and 32) by monitoring cell density, viability, viral infectivity, polyhedra production at 48,72,96 hrs post infection. Stabilized virus gave a better productihvity until late passages (passage 11-15) but did not sustain it to later passages.
Infectivity/DNA: A novel assay was designed to quantify the total viral DNA in a high throughput format by fluorescent staining of DNA and measuring the fluorescence in a fluorometer with plate reader. Using this assay, the specific infectivity (infectivity/DNA) of the passaged virus was measured to observe the effect of passaging on virulence. Very late passaged virus was found to have almost 20 times less infectivity/DNA than the early passaged virus for both the WT and stabilized viruses.
PCR analysis: Polymerase chain reaction (PCR) was performed to amplify the fp25k region to detect the host DNA insertions. No evidence of insertion was found in the fp25k gene at 11th, 15th, 26th and 32nd passage for both the WT and stabilized virus. Further analysis is underway to check for the mutations through selection of FP mutants and sequencing of the fp25k gene.
Transmission electron microscopy (TEM) of budded virus: The budded virus was precipitated from early and late passages and a size analysis was performed using a transmission electron microscope. A dramatic change was observed in the geometric size distribution of WT and stabilized virus from very late passage compared to that from early passage.
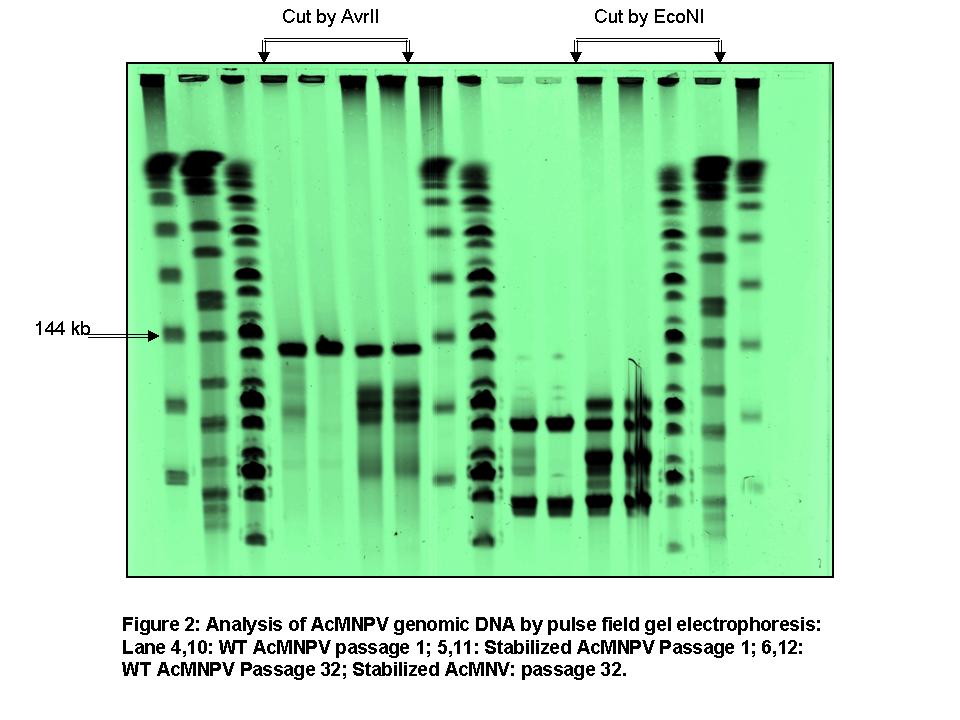
Pulse field gel electrophoresis of viral DNA and southern blotting (Fig 2): Pulse field gel electrophoresis was performed to identify the DIP formation and characterize the genomic size distribution of the early and late passaged viruses. It was found that there are four distinct genome-sizes present other than the WT genome-size all having deleted sequences. Southern blotting of these DNA (WT virus DNA as the probe) revealed that almost 50% of the viruses had deletion of large amount of DNA from their genome during passaging. For further confirmation of DIP formation, a restriction enzyme (EcoNI, which cuts the viral genome three times) analysis was performed with the passaged viral DNA.
Conclusion: We demonstrated that the removal of the transposon target sites from the wild type baculovirus fp25k gene (stabilized virus) reduced the incidence of the FP phenotype in late passages (passage 15), but did not eliminate the FP phenotype in very late passages (passage 30). Further, Genotypic and phenotypic analysis of late passaged virus showed that deletion of genomic sequences also contributed to the reduced infectivity and productivity. Production of DIP's with deleted sequences was shown for both WT and stabilized virus by characterization of the geometric size distribution and DNA size distribution of early and late passaged virus. Further, we corrected a commonly-held assumption (Kool et al.,1991) that FP mutations are precursor to DIP mutations, and showed that DIP mutations sometimes arise on their own. Hence, removal of transposon target sites from AcMNPV fp25k delayed incidence of the FP phenotype but had no impact on DIP production in cell culture.
Future plan: A two-stage continuous bioreactor (1.5L) set up is underway with controlled pH, oxygen concentration and temperature to evaluate the effectiveness of the stabilized virus. The quality and mutation (FP and DIP) pattern of the passaged stabilized virus with passaged WT virus. Specifically, The FP mutation will be identified by western blotting and Immunofluoroscence assay and DIP mutation will be identified by TEM and pulse field gel electrophoresis.
References:
Fraser, M.J.; Smith, G.E.; Summers, M.D. (1983). J Virol. 47(2): 287-300.
Kool, M.; Voncken, J.W.; Van Lier, F.L.J.; Tramper, J.; Vlak, J.M. (1991). Virology 183: 739-746.